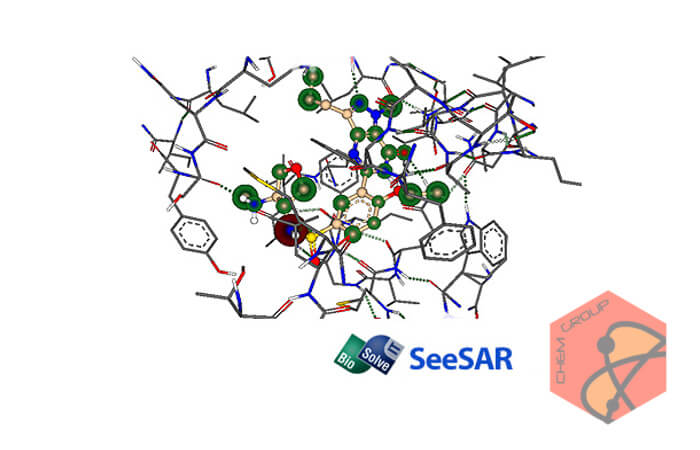
نرم افزار SeeSAR ابزاری برای شبیه سازی وارزیابی مواد مرکب ترکیبات مولکولی و اتمی است. این نرم افزار در فرآیند طراحی ساختارها از بهینه سازی چندپارامتری پشتیبانی می کند و همین امر باعث می شود تا ساختارهای طراحی شده تا حد بسیار زیادی به واقعیت نزدیک باشند.
امکان درنظرگرفتن پارامترهای مربوط به هر ترکیب در این نرم افزار، به علاوه ی امکان ایجاد طراحی های بلادرنگ سه بعدی، از SeeSAR یک دستیار کامپیوتری قدرتمند ساخته است.
قابلیت های کلیدی نرم افزار SeeSAR:
پیاده سازی گرافیک های پیچیده اتمی و مولکولی
برآورد تقریبی ΔS / ΔH- پشتبانی از پارامترهای فیزیکی و شیمیایی
وارد کردن داده های PDB یا Protein Data Bank
تجزیه و تحلیل نوسانات پیچشی گرما
SeeSAR is a software tool for interactive, visual compound prioritization as well as compound evolution. Structure-based design work ideally supports a multi-parameter optimization to maximize the likelihood of success, rather than affinity alone. Having the relevant parameters at hand in combination with real-time visual computer assistance in 3D is one of the strengths of SeeSAR.
Affinities:
We implemented sophisticated graphics to visualize atom-based affinity contributions; that allow for a rough estimate of the ΔS / ΔH -split of the Free Energy. (This is an ongoing co-development between BAYER, the University of Hamburg and BioSolveIT.)
Phys-chem properties:
Relevant parameters are computed on-the-fly or imported to be taken into consideration throughout the design process.
Torsional 'heat':
Torsional statistics analyses (developed between Hoffmann-LaRoche and the University of Hamburg); is readily available via intuitive color-coding.
Explorable space:
A tight fit is the prerequisite for both, affinity and specificity. Therefore, as guidance for the user, efficient computation combined with refined graphics provides on-the-fly visualization of gaps in the binding interface and positions where a tighter fit is likely to be gained.
2D molecule browsing - time to look at things from a different angle!
While the molecule table offers great functionality for prioritizing compounds based on the data, it does not provide an overview of the molecules themselves. This release, however, sees the introduction of 2D molecule browsing. The table now offers two views - the one you already know and a 2D browser - flick between them using the switch below the table. Both views are always kept in sync so if you add a filter or sort etc. the 2D browser will show you the same result in the same order as the table. Also try expanding the table area to see how more molecules fit into the view.
Fantastic new 3D graphics features
This release also brings with it some great new 3D graphics improvements. As much as we all like visualising the binding site surface, it lay often times in the way… The binding site surface can now be switched to transparent allowing you to see through it and therefore making the analysis of the binding site and molecules within much more comfortable. Also, the feeling of depth in the 3D view has been improved to help orientation - a so-called "fog effect" fades out the protein and molecules that are further away to bring the foreground more into focus.
Persistent amino acid labels and better view of reference
So far, labels on binding site components unfortunately disappeared when browsing through different molecules in the table. Now amino acid, co-factor and water labels remain present if you change to a different molecule in the 3D view and even if you enter the molecule editor. The view of the reference compound has also been improved. For better visibility, the thickness of the bonds has been increased and instead of coloring the whole molecule in a uniform blue color, only the carbon atoms are colored blue so that hetero atoms can be distinguished more easily.
1- نرم افزار را نصب کنید.
2- نرم افزار را اجرا نکنید و اگر در کنار ساعت نیز در حالت اجرا قرار داد آن را ببندید.
3- محتویات پوشه Crack (فایل های biosolveit.lic و Patch.exe) را در محل نصب نرم افزار* کپی کنید و فایل Patch.exe را اجرا و عملیات Patch را انجام دهید. (توجه داشته باشید چنانچه از ویندوز های 7 و 8 و یا 10 استفاده می کنید برای اجرای فایل Patch.exe می بایستی بر روی آن راست کلیک کرده و گزینه Run as administrator را انتخاب کنید تا Patch به درستی کار کند)
4- نرم افزار را اجرا کنید.
* محل نصب نرم افزار: پوشه محل نصب معمولاً در درایو ویندوز و داخل پوشه Program Files قرار دارد. همچنین با این روش می توانید محل نصب را پیدا کنید:
در ویندوز XP: بعد از نصب، روی Shortcut نرم افزار در منوی Start کلیک راست کرده و روی گزینه Properties و سپس روی گزینه Find Target کلیک کنید.
در ویندوز 7: بعد از نصب، روی Shortcut نرم افزار در منوی Start کلیک راست کرده و روی گزینه Open file location کلیک کنید.
در ویندوز 8: بعد از نصب، روی Shortcut نرم افزار در صفحه Start Screen کلیک راست کرده و روی گزینه Open file location کلیک کنید، در پنجره ایی که باز می شود مجدداً روی Shortcut نرم افزار کلیک راست کنید و روی گزینه Open file location کلیک کنید.
در ویندوز 10: بعد از نصب، روی Shortcut نرم افزار در منوی Start کلیک راست کرده و روی گزینه Open file location کلیک کنید، در پنجره ایی که باز می شود مجدداً روی Shortcut نرم افزار کلیک راست کنید و روی گزینه Open file location کلیک کنید.
لینک دانلود شبیه سازی ترکیبات مولکولی و اتمی SeeSAR v5.4
حجم فایل: 25.4 مگابایت
شرکت سازنده: BioSolveIT GmbH
پسورد: www.chemgroup.ir

برای نوشتن دیدگاه وارد حساب کاربری خود شوید.